



Search websites, locations, and people

MeDuSA: a novel method for investigating cellular state dynamics in tissue-level transcriptomic data
Jian Yang Lab
14, 2023
Email: zhangchi@westlake.edu.cn
Phone: +86-(0)571-86886861
Office of Public Affairs
Most of us have likely undergone routine blood tests due to physical discomfort. These tests assist doctors in diagnosing our ailments and formulating corresponding treatment plans. For instance, when bacteria infect our upper respiratory tract, we may observe an increased proportion of neutrophils in our blood. Activated neutrophils, acting as the "immune police", help our bodies clear invading bacteria. However, current routine blood tests in hospitals can only inform us of an increase in the number of neutrophils, without specifically indicating how many of these neutrophils are in an activated state. Indeed, there are many different types of cells in our bodies that can dynamically change their states under various physiological or pathological conditions. Furthermore, when dealing with complex solid tissue diseases, such as malignant tumors, is there a way to determine which cells in the tissue have changed their functional states and what their proportions are?
On July 13, 2023, Nature Computational Science published online a research study led by Liyang Song, a doctoral student from Prof. Jian Yang’s lab at Westlake University, titled “Mixed model-based deconvolution of cell-state abundances (MeDuSA) along a one-dimensional trajectory". This study developed a method named MeDuSA to estimate the abundances of cells at different states in tissue-level transcriptomic data. The research team applied this method to various diseases including human cancers and COVID-19, successfully identifying cell states related to disease onset, development, prognosis, and drug treatment responses. This work significantly enhances our understanding of cell state dynamics in different biological processes.
 https://www.nature.com/articles/s43588-023-00487-2
https://www.nature.com/articles/s43588-023-00487-2
With the rapid development of single-cell transcriptome sequencing technology (scRNA-seq), we are now able to obtain high-throughput transcriptomes of cells in different states within tissues. By integrating computational analysis methods, we can use single-cell transcriptome information to infer the states of individual cells and explore changes in cell states during disease. However, due to the high cost of single-cell technology and stringent quality requirements for biological samples, the widespread application of scRNA-seq technology in large-scale biological samples remains a challenge. Compared to scRNA-seq, tissue-level transcriptome sequencing (bulk RNA-seq) has the advantages of lower cost and less demanding tissue quality requirements, making it more suitable for use in large-scale biological databases. However, because bulk RNA-seq lacks single-cell level information, we cannot directly observe changes in cell states through transcriptomes. So, could we combine the advantages of these two sequencing technologies, use scRNA-seq data generated from a limited number of samples as a reference, and then infer cell state transitions based on bulk RNA-seq data? This approach would allow us to study the relationship between cell states and diseases on the scale of large biological samples.
The MeDuSA method developed in this study is based on such a cellular deconvolution strategy. It utilizes scRNA-seq data from small samples as a reference to estimate the abundances of cells in different states in bulk RNA-seq data. Many previously established cell deconvolution methods, due to limitations in model architecture, could only infer cell-type abundance (such as neutrophils), but were unable to accurately estimate cell-state abundance (such as activated neutrophils). The MeDuSA method employs mixed linear models to fit the transcriptome of individual cells, systematically reducing model residuals and optimizing estimation bias caused by strong correlations between cell states, thus accurately estimating the abundance of cell states. This analysis method, combining scRNA-seq and bulk RNA-seq, overcomes the bottleneck of studying cell states in large-scale biological databases, taking a critical step towards revealing the cell state dynamics behind diseases and their related biological mechanisms.
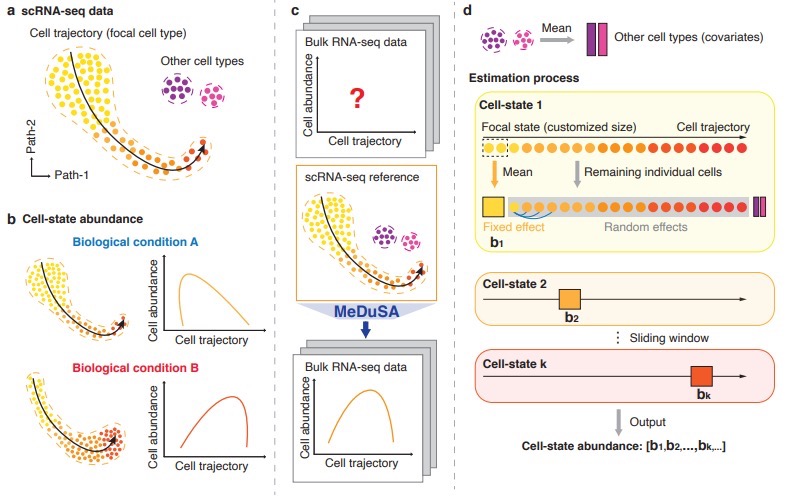
Figure 1. Conceptual diagram of the MeDuSA method
The authors used MeDuSA to perform a detailed analysis of bulk RNA-seq data from esophageal tissues from 710 donors. They found that, compared to normal esophageal tissue, the epithelial cells in the tissues of patients with esophageal tumors tended to be significantly biased towards the basal layer (proliferative layer). This finding aligns with the histological understanding that esophageal tumors mainly originate from the basal layer of the esophagus. The authors also analyzed data from the peripheral blood of 215 donors and found that, compared to healthy donors, the abundance of activated CD8+ T cells in the blood of COVID-19 patients was significantly increased. Furthermore, the abundance of activated CD8+ T cells was positively correlated with the severity of COVID-19 infection. In analyzing data from 507 melanoma patients, the authors found that most of the CD8+ T cells in these patients' tumor tissues were in an exhaustion state, and the proportion of cells in this exhausted state was positively associated with the degree of T cell receptor expansion. Notably, they also discovered a strong association between the exhaustion state of CD8+ T cells and the survival time, as well as the immune treatment response of melanoma patients. These results suggest that the state of CD8+ T cells could serve as an important biomarker, with significant implications for clinical decisions regarding the use of immunotherapy. In addition, by combining whole-genome sequencing data, the authors identified 162 genes with epithelial cell differentiation-dependent expression quantitative trait loci (eQTL), indicating that cell states play an important role in genetic regulation.
The study also outlined potential future research directions, suggesting that as spatial transcriptomics becomes more prevalent, the opportunity will arise to use spatial transcriptomics as a reference to infer the distribution of spatial cell states from bulk RNA-seq data. This research direction will further deepen our understanding of the cell state dynamics behind diseases, thereby opening new avenues for future disease prevention and treatment.
The MeDuSA method has been integrated into a user-friendly analysis software and has been open-sourced at the following link: https://github.com/LeonSong1995/MeDuSA.
This research was supported by the National Natural Science Foundation, Westlake Laboratory, Westlake Education Foundation, and the Research Center for Industries of the Future and High-Performance Computing Center of Westlake University.
RELATED














-
STUDY WITH US
-
WORK WITH US
-
CONTACT US
-
YUNGU CAMPUS
-
No. 600 Dunyu Road, Sandun Town, Xihu District, Hangzhou, Zhejiang PR China Post code:310030 E-mail:office@westlake.edu.cn
-
-
YUNQI CAMPUS
-
No.18 Shilongshan Road Cloud Town, Xihu District, Hangzhou, Zhejiang PR China Post code:310024 E-mail:office@westlake.edu.cn
-
-
-
浙ICP备18025489号 浙公安备33010602007514号 Copyright © Westlake University. All Rights Reserved
-


If you are interested in supporting us or making a contribution, please contact:
Office of Public Affairs, Westlake University
Email: media@westlake.edu.cn
OR
Westlake Education Foundation
Email: office@wefoundation.org.cn
Phones: 0571-86886859
Fax: 0571-85271986